Primer hiperoksalüri tip 1 (PHI)
Primer hiper okzalürisi olan hastada oksalat için endojen kaynak aşağıdakilerden hangisidir?
a. Glutamin
b. Alanin
c. Glisin
d. Lösin
e. Metyonin
Parankimal dokularda kalsiyum oksalatın depolanması "oksalozis" olarak bilinmektedir. Hiperoksalüri kalsiyum oksalatın idrarda süpersatürasyonu nedeniyle nefrolitiyazis veya nefrokalsinozis ile karakterizedir. Hiperoksalürinin başlıca primer, sekonder ve idiyopatik olmak üzere 3 tipi mevcuttur.
Primer hiperoksalüri tip I, II ve III karaciğerde glioksalat metabolizmasındaki bir defekt nedeni ile oluşan otozomal resesif geçişli hastalıklardır.
Sekonder hiperoksalüri böbrek fonksiyonlarındaki bozukluktan dolayı oksalatın atılımındaki azalma veya askorbik asit, methofluran, etilen glikol ve xylitol alımına bağlı olarak aşırı oksalat alımı veya kronik inflamatuar bağırsak hastalığı, intestinal bypass, ince bağırsak rezeksiyonu ve eksternal safra drenajı gibi oksalatın absorbsiyonunda artmaya bağlı olarak gelişmektedir.
İdiopatik oksaloziste bilinen bir gen defekti saptanmamıştır. Olası sebeplerinin genetik olarak oksalatın fazla üretimi veya oksalatın membran transportundaki anormalliklerden dolayı olabileceği düşünülmektedir.
Tedavi edilmediğinde hiperoksalüri, böbrek yetmezliğine ve böbrek dışı dokularda da birikerek oksalozise neden olmaktadır. Oksalat depolanması sıklıkla kemik, kemik iliği, kan damarları, kalp, santral sinir sistemi, periferal sinirler, retina, deri ve tiroid dokusunda olmaktadır.

Primer hiperoksalüri tip 1 (PHI) peroksizomal alanin gliyoksilat aminotransferazin (AGT) karaciğer hücrelerindeki eksikliğine veya AGT'nin mitokondriyumda bulunmasına bağlıdır. Nadir görülen, gliyoksilat metabolizmasının otozomal resesif kalıtılan bir bozukluğudur
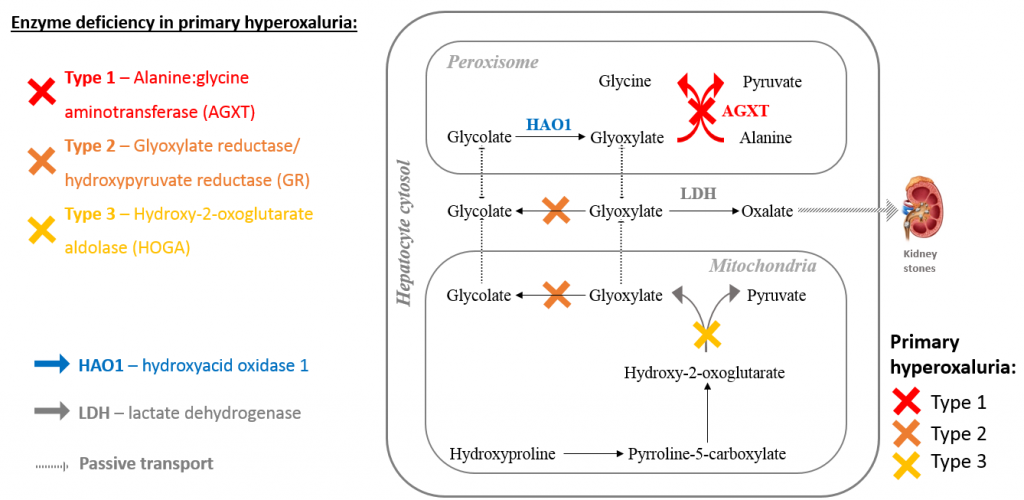
AGT karaciğer hücrelerinde gliyoksilatın glisine transaminasyonunu katalize eder. Gliyoksilatın aminasyon yetersizliği sonucunda peroksizomda gliyoksilat oksidaz veya sitozolde laktat dehidrogenaz enzimi ile aşırı oksalat oluşur. Artmış renal oksalat atılımı ile idrarda kalsiyum oksalat kristalleri meydana gelir, bunların çözünürlüğü düşüktür. Tekrarlayan üriner sistem taşları, nefrokalsinozis ve progresif renal yetmezlik ortaya çıkar Renal fonksiyon yetersizliği belirdiğinde, yalnızca böbrekten atılabilen oksalat kemikler, kaslar, arterler, gözler, cilt, sinirler ve diğer dokularda birikir. Bu tabloya oksalozis denir ve çeşitli klinik belirtilere yol açar

Primer hiperoksalüri tip 2'de (PH2) ise 87 karaciğerde D-gliserat dehidrogenaz enzimi eksiktir; bu enzim gliyoksilat redüktaz ile özdeş olup, hiperoksalürinin yanında L-gliserik asidüriye neden olur. PHl'e göre çok nadirdir ve hiperoksalürili hastaların %9-20'sinde görülür. İdrar oksalat düzeyi daha düşüktür, sistemik oksalozis daha nadir görülür.
Hyperoxaluria; Bird's disease olarak da bilinir.
Type I primary hyperoxaluria (PH1) AGXT “Serine—pyruvate aminotransferase” gen mutasyonu ile ilişkilidir
Type II ise GRHPR “Glyoxylate reductase/hydroxypyruvate reductase” ile ilişkilidir.
Secondary hyperoxaluria jejunoileal bypass, kuzukulağı tüketimine bağlı oluşur.